The Zymo-Seq RiboFree™ total RNA-seq library kit from Zymo Research is the fastest and easiest RNA-seq library preparation kit available to produce stranded, total RNA libraries. Featuring an automation-friendly protocol for minimal hands-on time, this kit generates sequencer-ready, stranded, indexed libraries depleted of rRNA and globin in as little as 3.5 hours.
The probe-free depletion developed by Zymo Research uses only abundant pre-existing transcripts for mismatch-free enzymatic removal of rRNA and globin, resulting in minimal off-target effects as low as 1.8% in all protein-coding transcripts.
Benefits of using the Zymo-Seq RiboFree total RNA-seq library kit
• 3.5 hour, fragmentation free protocol
• Compatible with any organism
• Compatible with any biological sample
• Probe free
• Input 100ng - 2ug total RNA or 50ng ribosome depleted RNA samples
• Pre-mixed reagents, indexing primers, SPRI beads and magnetic bead stand included
The fastest RNA-Seq library prep kit
RiboFree universal depletion (rRNA, betaglobin): probe-free and compatible with all biological samples types
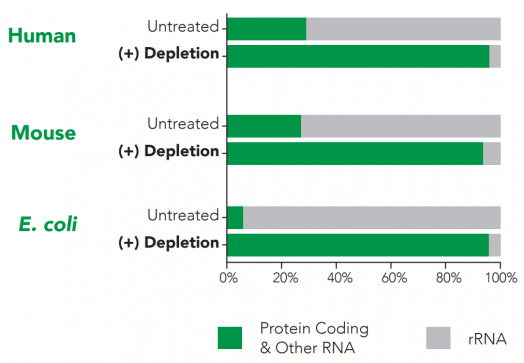
RiboFree universal depletion will enzymatically remove rRNA from any sample type. Paired end sequencing was performed on stranded total RNA-Seq libraries, both with and without RiboFree universal depletion. Read pairs were aligned to their respective genomes using the STAR aligner. Read classes were defined using a combination of Ensembl GTF gene biotypes and RepBase repeat masker annotations. Number of reads overlapping each annotation class were divided by total reads in that library to calculate percent reads of each annotation class.
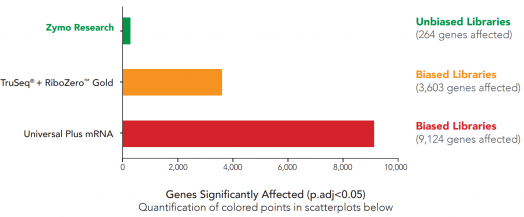
Probe-free technology eliminates bias: 35x less biased expression profiles
![RiboFree™ Universal Depletion maintains native expression profiles unlike TruSeq® Total RNA [probe-based Ribo-Zero™ Gold] and Univeral Plus mRNA-Seq [poly(A) enrichment].](/userfiles/images/zymo_seq_unbiased_libraries_zymo_data.png)
![RiboFree™ Universal Depletion maintains native expression profiles unlike TruSeq® Total RNA [probe-based Ribo-Zero™ Gold] and Univeral Plus mRNA-Seq [poly(A) enrichment].](/userfiles/images/zymo_seq_unbiased_libraries_true_seq_data.png)
![RiboFree™ Universal Depletion maintains native expression profiles unlike TruSeq® Total RNA [probe-based Ribo-Zero™ Gold] and Univeral Plus mRNA-Seq [poly(A) enrichment].](/userfiles/images/zymo_seq_unbiased_libraries_universal_plus_data.png)
RiboFree Universal Depletion maintains native expression profiles unlike TruSeq® Total RNA [probe-based Ribo-Zero™ Gold] and Univeral Plus mRNA-Seq [poly(A) enrichment]. Paired-end sequencing was performed on libraries prepared from Universal Human Reference RNA (Invitrogen) containing ERCC Spike-In Mix 1 (Life Technologies), both with and without rRNA removal or poly(A) enrichment. Libraries were sequenced to a depth of ~35 million reads per library, and read pairs were aligned to the hg38 human genome using the STAR aligner. Read classes were defined using Ensembl GTF gene biotypes. The DESeq2 package was used to apply the “apeglm” log-fold-change shrinkage estimator to determine which of the 20,004 protein coding genes and ERCC Spike-In transcripts were significantly affected (p.adj < 0.05) by rRNA removal. Significantly affected transcripts are represented as coloured points in the scatterplots.
Material available for download
Zymo-Seq RiboFree total RNA library kit manual (R3000S)
Zymo-Seq RiboFree total RNA library kit manual (R3000)
Zymo-Seq RiboFree universal cDNA library kit manual
TruSeq is a registered trademark of Illumina Inc.
Products
Note: product availability depends on country - see product detail page.
| Details | Cat number & supplier | Size | Price |
| Zymo-Seq RiboFree Total RNA Library Kit (12 preps) R3000 · Zymo Research | R3000 Zymo Research |
12 preps |
£1317.00
12 preps
view
|
